
Check Out Our New Forum!
The Your Turn Forum provides a space where people can post questions about mesh and medical device related issues and get advice and support from the Mesh News Desk community.
Join the Discussion!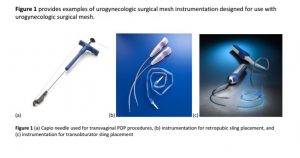
Surgical tools, FDA
Mesh Medical Device News Desk, January 16, 2017~ Pelvic mesh tools used to place and affix transvaginal mesh had been a low-risk FDA category alongside Band-aids. Because of injuries, it's been placed in a class II or moderate-risk category.
Tools used to place transvaginal mesh have been found to cause injuries in up to 25% of pelvic mesh placements.
The surgical instruments include needle passers and trocars or stainless steel hooks. There are also fixation tools to affix the pelvic mesh to the tissues and anchors that are implanted deep into ligaments making them impossible to remove.
The U.S. Food and Drug Administration (FDA) didn’t think these tools presented much of a risk and had them share a class I low-risk category with Band-Aids and wheelchairs. But data shows otherwise.

Perigee System, superior and inferior needles
The MAUDE database of the FDA, that records adverse events, shows hemorrhage, bleeding, organ perforation, nerve injury and pain are all related to the use of this specialized instrumentation used in a “blind” procedure to place mesh.
That means the doctor cannot directly visualize placement of surgical mesh and must rely on tools and feel despite the fact that they pass near nerves, tissues and organs.
On January 6, 2017, the FDA reclassified these tools to moderate risk or class II, from low risk or class I. Some public comments in the Federal Register had suggested it be made to a class III high risk.
In making that recommendation, the FDA had searched its Medical Device Report (MDR) database from January 2008 to December 2015. In all it found 463 MDRs, with the most of them submitted by manufacturers, 14 by a user facility and 11 voluntary reports.
Interestingly, “The FDA then removed reports that included the terms attorney and plaintiff, as such reports typically contain few details and stem from patient litigation.”
Boston Scientific and Ethicon were the two manufacturers with the highest number of instrument malfunction problems including suture and needle detaching or breaking. Boston Scientific had 316 reports while Ethicon (Johnson & Johnson) had 90, the two highest number of adverse event reports.
Boston Scientific’s Pinnacle Pelvic Floor Repair Kit had the highest number of adverse event reports at 134 and Uphold Vaginal Support System at 167.

The reclassification of mesh surgical tools was put in the Federal Register in May 2014, the same time the FDA published a proposed order to reclassify the larger surgical mesh for pelvic organ prolapse (POP) from class II to class III or high risk.
POP mesh was reclassified beginning in January of 2016 to high–risk class III but mesh makers were given 30 months to either prove their mesh was safe and effective or stop selling it.
Just like transvaginal mesh, which is put into the moderate risk class II even though it is plastic permanently implanted in the body, the FDA had underestimated the amount of harm that pelvic mesh and its tools could cause.
Some consumer comments to the proposal suggested the surgical tools share the high risk category with pelvic organ prolapse, (POP) mesh, a larger polypropylene mesh support system.
Reclassifying requires tool manufacturers must demonstrate a reasonable assurance that the surgical instruments are safe and effective, biocompatible, sterile, and packaged correctly and safety and function as intended. Manufacturers will be required to perform tests on the instruments and label them accurately regarding their expiration date, methods and instructions, sterilization and mesh design that’s compatible with the device.
Instead, most POP mesh has already been quietly removed from the market by manufacturers, eager to avoid the future cost of thousands of defective product lawsuits. ###




